Spatial omics 3 – proteomics with immunolabelling

In this entry, we delve into the OG spatial proteomics technique - immunohistochemistry.
This post is part of our series titled "Introduction to spatial omics", which contains the following entries:
- Spatial omics part 1: the history and how of histology
- Spatial omics 2 – Mixing Modern Methods mit Microscopy
- Spatial omics 3 – proteomics with immunolabelling (current post)
- Spatial omics 4 - Antibody visualization: from microscopy to mass spectrometry
Table of contents
Blog post by request! Since immunohistochemistry is arguably the first spatial proteomics method, this entry explains some of the underlying principles of this technique.
Introduction to immunohistochemistry.
Continuing with our series on spatial omics for tissue analysis, it’s time to expand a bit more on immunohistochemistry and immunofluorescence – I’m just going to refer to both as IHC. When you think of IHC, you’re probably thinking of blue and brown pictures like this:
And you wouldn’t be wrong. However, the technique that underpins IHC also produces images like this:
And this:
because techniques like multiplexed immunofluorescence and Imaging Mass Cytometry also use antibody labelling 1. While stains such as H&E are non-specific, antibody labelling such as in IHC allows users to visualize the distribution of a target protein or proteins in a tissue context. This can be used to look at their location and colocalization within cells, investigate things such as specific cell subpopulations, changes in protein expression or cell morphology, and of course in the diagnosis of diseases.
Definition of terms
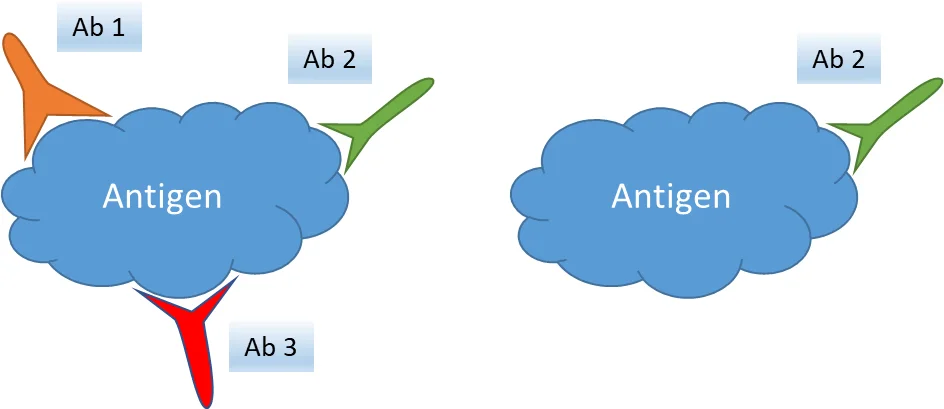
Whether it’s single marker chromogenic IHC, multiplexed immunofluorescence or mass cytometry imaging, proteins are labelled via antibody-antigen interactions. Again, let’s define some terms
- An antigen (Ag) is a molecule or structure against which an immune response is produced – for example, the SARS-COV2 spike protein – against which an antibody (Ab; a serum protein also known as an immunoglobulin) is raised. An epitope is a specific part of the antigen that the antibody binds to. An antigen can have many epitopes, meaning that potentially multiple antibodies can bind to it.
- Antibodies are generated by plasma cells (activated B lymphocytes) and can be monoclonal or polyclonal.
- A single plasma cell will produce antibodies that bind to only one epitope. Monoclonal Abs are generated by fusing a plasma cell that produces a specific antibody to an immortal B cell line using science that’s too complex to explain in a few lines (I tried), so that it can produce the same antibody clone repeatedly in unlimited quantities in labs. Monoclonal Abs are homogenous, and highly specific as they bind to a single epitope on the antigen.
- Polyclonal Abs are generated by immunizing an animal (e.g. a rabbit or mouse) against a specific antigen. After the animal produces an immune response against the antigen, Abs can be collected from their serum. Polyclonal antibodies are so named because they contain a mixture of different Abs – multiple plasma cells will have produced antibodies against different epitopes on the antigen. Polyclonal Abs are thus more sensitive than monoclonal Abs as the multitude of Abs against multiple epitopes means binding is more likely to occur when applied to a sample.
- IHC can be performed with either a direct or indirect mechanism. For the direct method, the Ab that detects the antigen (primary Ab) has a label (e.g. a fluorophore) directly attached to it. For the indirect method, the primary antibody is not labelled – instead, the primary antibody is detected by a secondary Ab which itself has the label.
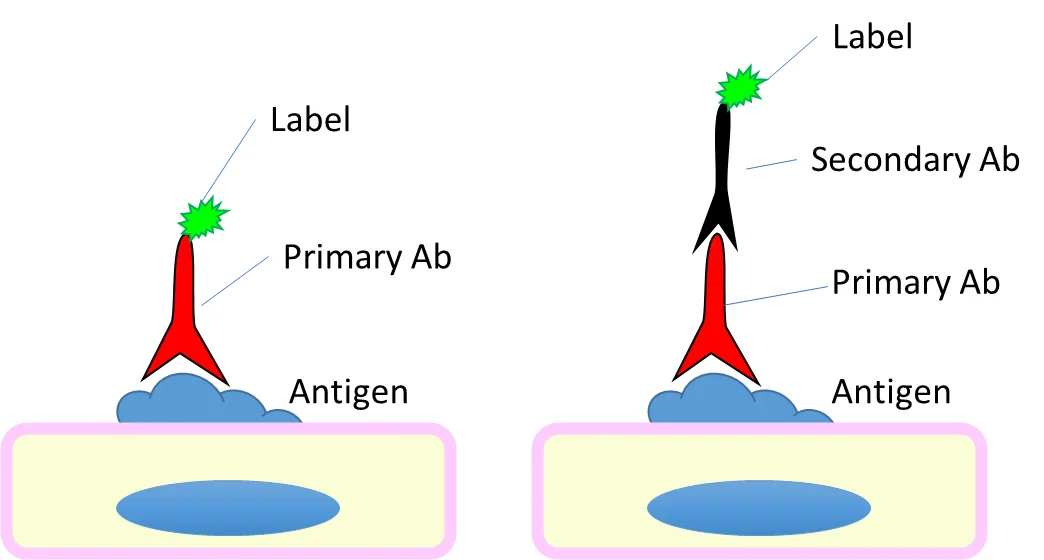
- Antibodies are conjugated to a label that allows us to visualize them and extrapolate the location of their antigen.
a. Enzyme chromogens use an enzymatic reaction to produce a stable, colored precipitate. A frequently used chromogen combination is horseradish peroxidase (HRP) with diaminobenzidine (DAB) substrate which produces brown staining such as what is seen in the first tweet above. As IHC using chromogenic detection can be viewed using a standard light microscope and is stable enough to be stored for years, it is commonly used in diagnostics.
b. Reading over my textbooks for this blog, I was a bit shocked to learn that antibody labelling with fluorescence was reported EIGHTY YEARS AGO by Coons et al, in 1941 2?! Fluorescent probes absorb light at a certain wavelength and then emit light at another wavelength, typically in the visual spectrum. There are many commercially available fluorescence probes in all the colors of the rainbow, and can be viewed on fluorescence-enabled microscopes.
c. Oligonucleotide conjugation of antibodies is becoming increasingly common, especially for multiplexing purposes 3 ,4. In this case, detection is conducted by hybridizing the oligo sequence with a complementary chain that is attached to a fluorescent probe. This method allows for multiple hybridization and signal removal rounds.
IHC protocol outline
The following is an example of the major steps for indirect fluorescent IHC of FFPE sections after the sections have been mounted and annealed onto microscopy slides. This would of course have some steps omitted or modified as required for a different type of IHC, for example direct IHC with an enzyme chromogen.
- Deparaffinization and rehydration Paraffin is removed from the sections by washing in xylenes, rehydrated in graded alcohol solutions, and washed in water. This step is very important as paraffin is hydrophobic and can repel different buffers and solutions.
- Antigen retrieval and washingThis step is used to break formalin crosslinks between proteins in the tissues formed during tissue fixation. There are different possible methods for this, depending on the antigen, such as heating the sample in a buffer or using acid. The slides should be washed in a buffer (e.g. phosphate buffered saline) to remove residual antigen retrieval washing solution. For FFPE sections, permeabilization of the sections can be conducted by adding a detergent (e.g. Triton X-100) to the washing buffer. This is particularly useful for intracellular proteins. After these steps, if the IHC is being conducted manually, the samples are circled using a hydrophobic pen to keep the following reagents on the sections.
- Blocking Tissues contain endogenous antibodies that might lead to non-specific binding or high background signals. Incubating the sections using normal serum from the same species that the secondary Ab is derived from can be used to ‘block’ these signals. For example, goat serum is used if the secondary is a goat antibody.
- Primary Ab incubation and washing The tissue sections are covered with primary Ab, allowing binding to happen if the antigen is present. Incubation time varies depending on the Ab, but typical durations are 1-2 hours at room temperature or overnight in the fridge. Excess unbound Ab is washed away with repeated cycles of washing in a buffer.
- Secondary Ab incubation and washing The sections are covered with secondary Ab (1-2 hours at room temperature), followed by repeated buffer washes. For fluorescent IHC, incubation of the secondary Ab should take place in the dark (e.g., in a covered box or cupboard) as light exposure will cause fading of the conjugated fluorescent probes.
- Counterstaining, mounting and coverslipping As IHC should only show antigens of interest, counterstains are used to provide tissue context. These are typically stains that label cell nuclei, such as haematoxylin, DAPI or SYTOX Green. In fluorescence, it is important to select a counterstain that will emit a signal that can be separated from the secondary Ab, e.g., if the secondary Ab is conjugated to a green fluorescent tag, DAPI which emits in the blue range is suitable whereas SYTOX Green might not be distinguishable. After finishing the staining, the sections are typically covered with a thin glass coverslip to prevent dehydration and protect the sample. Some mounting solutions contain anti-fade agents and DNA stains, removing the need for a separate counterstaining step.
The next step is viewing your sample via microscopy, or if you’re really fancy, via Imaging Mass Cytometry (IMC) or Multiplexed Ion Beam Imaging (MIBI), but I think that’s something for another blog entry.
Summary
This blog covered some of the technical background and a basic protocol for fluorescent IHC. If you’re looking for some tips on IHC image and other spatial omics analysis, contact Aspect Analytics for ideas.
References:
- Hickey JW, Neumann EK, Radtke AJ, et al. Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat Methods 2021.
- Bancroft's Theory and Practice of Histological Techniques. 8th ed. Elsevier: 2019.
- Goltsev Y, Samusik N, Kennedy-Darling J, et al. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018;174:968-81 e15.
- Jones JA, McMahon NP, Zheng T, et al. Oligonucleotide conjugated antibody strategies for cyclic immunostaining. Sci Rep 2021;11:23844.